I tried to run the latest VFVS-2 branch with slurm. After submitting jobs, I get a FileNotFoundError when using an older VF1 ligand library (from Enamine-REAL-2018q12_isomers.pdbqt).
Has anyone successfully run the VFVS-2 code with slurm and the REAL-2018q12 tranches? I don’t have access to the real-space-2022q12 libraries yet, that is why I am trying with slurm and the older libraries.
Any help is much appreciated. Thanks!
Oliv
Error in /VirtualFlow/VFVS-vfvs-2/workflow/workunits/1/803_0.err:
File “/VirtualFlow/VFVS-vfvs-2/tools/./templates/vfvs_run.py”, line 157, in downloader
shutil.copyfile(Path(item[‘collection’][‘sharedfs_path’]), item[‘local_path’])
File “/miniconda3/lib/python3.10/shutil.py”, line 254, in copyfile
with open(src, ‘rb’) as fsrc:
FileNotFoundError: [Errno 2] No such file or directory: ‘/VirtualFlow/VFVS-vfvs-2/input-files/ligand-library/GACAAF/00000.tar.gz’
Hi Haroon,
No, I was not able to resolve it. I spent about one day debugging and rewriting code, then I gave up.
I decided to wait until VFVS-2 is fully baked and released which will hopefully happen soon.
Best, Oliv
Hello @oeidam ,
I was having similar issues, VFVS2 is finicky with relative file paths, I was able to get it to work by pointing it to a specific meta-tranche in collection_folder parameter of the all.ctrl file. My setting was to …/input-files/ligand-library/AB. I used it with a different dataset but it ran at least. I ran into many isses with setting file paths, and was only able to figure it out by going through the code and printing certain paths created by vfvs_run.py along the way to figure out what it wanted.
Also be aware that tar files (the format in which the ligands are contained) have a relative file path within them that is retained from the working directory of whatever script packaged them. So if they were processed with a path VFVS/input-library/A/… they would be extraced in the shared memory file sysstem with that same path which is not necessarily what Virtualflow expects. It was one issue I had.
Hi @Behrgen, thanks for the solution. Are you using AWS or local cluster? I have setup VFVS1 on my local single node cluster, I want to now run VFVS2 on my local cluster, is that possible or not, or is VFVS2 is only supported on cloud based clusters?
@oeidam please also answer if you have any idea about it.
Hi @har_vf ,
I run VFVS-2 successfully on my local cluster with SLURM. Input files are small ligand sets containing vflp-2 prepped molecules. Mols from the 1.4B Enamine REAL (2018q12) library also work with tweaks. I have not tried to access the big 69B library (Enamine_REAL_Space_2022q1) from slurm. For the big library, and for the ATG-VS-specific scripts of VFVS-2, I use the AWS Batch system. The most important thing is to follow the tipp from @Behrgen with regards to the ligand-library path in all.ctrl (collection_folder=…/input-files/ligand-library/AB instead of collection_folder=…/input-files/ligand-library).
In my hands, the vfvs-2 python scripts are a bit slower compared to vfvs-1 bash scripts on SLURM. And despite this I prefer the new python scripts because of readability.
HTH, Oliv
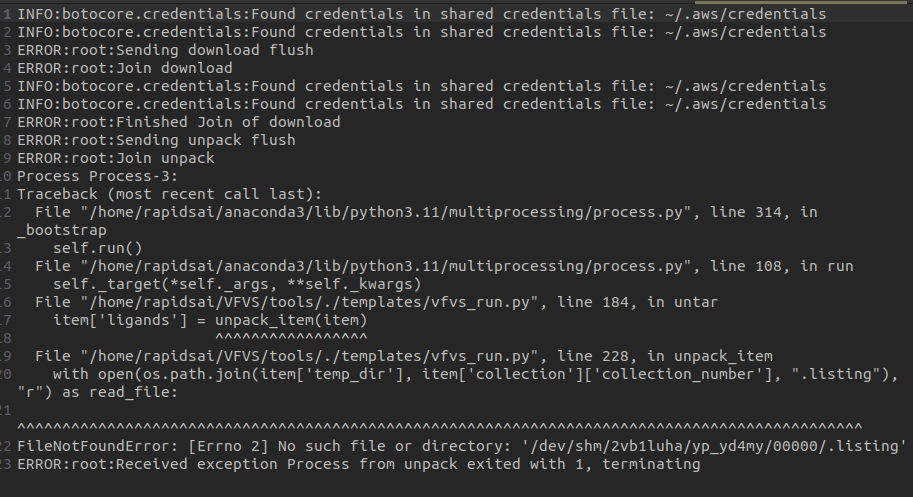
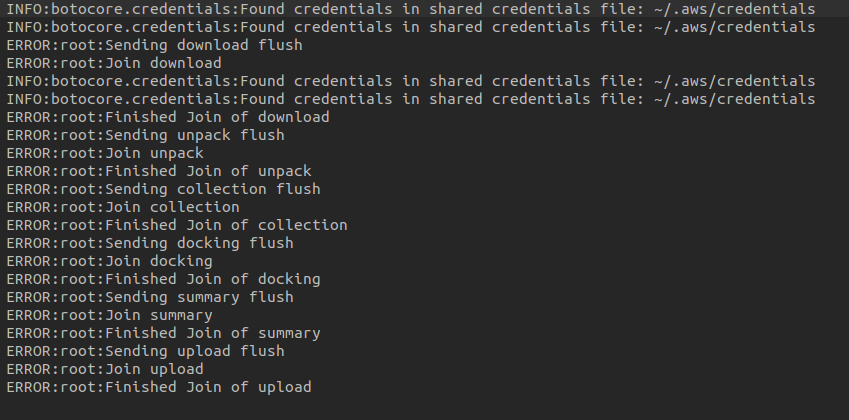
thanks @oeidam , i was able to resolve that issue and the the docking were performed. But VFVS-2 is still giving an error in the workunit folder of workflow directory. I have attached the picture of the error, please let me know how to resolve it.
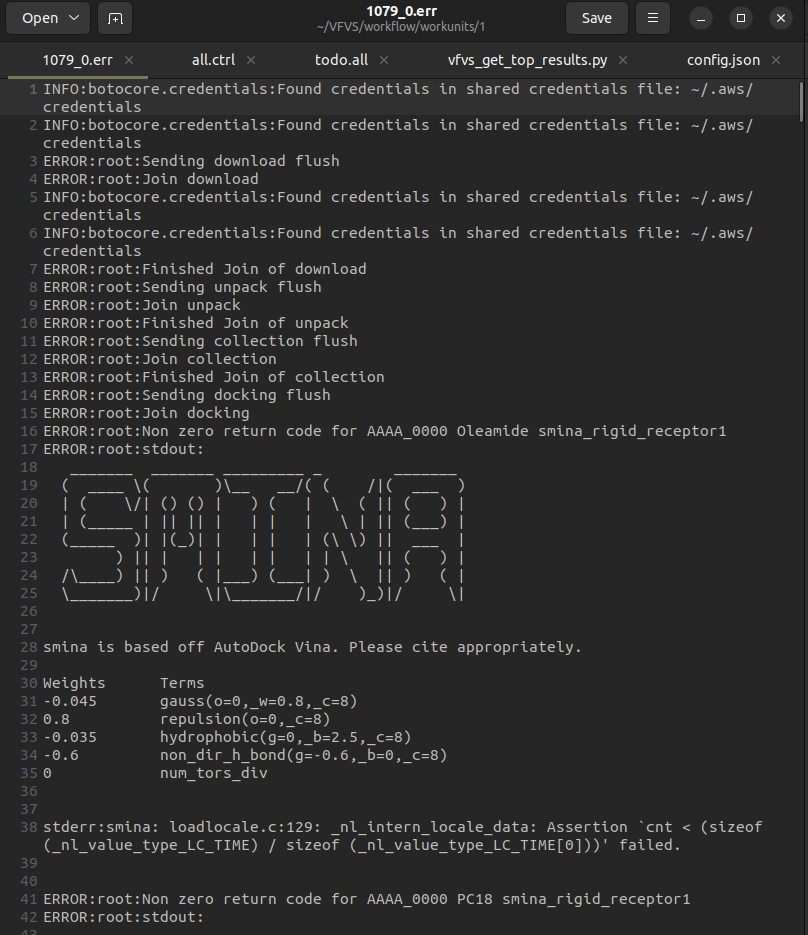
hey @oeidam , have you used smina docking program in VFVS2? I am able to run the docking with Qvina docking program (though with the previous error), but I am not able to run the docking with Smina docking program. The error it throws on running VFVS2 on Smina docking program is attached in a screenshot. Can you help me out in resolving it? Thanks
Howdy Haroon,
I had the same problem with smina initially.
To fix this problem, type the following in the shell: export LC_ALL=C
To make the fix permanent, add a new line to your ~/.bashrc with the above line.
Happy weekend, Oliv
hey @oeidam thanks for the reply, it solved the issue for me. But still some of the ligands are failing to dock, the reason for this failing is “timeout on ligand_name” in the overview.json file of output-files directory.
I changed the values of program_timeout and slurm_job_submission_timeout but it does not affect much. But when i run the docking on the same settings, it dock 2 3 more compounds on each run or some times less compounds, but this issue only comes on smina docking program, qvina is docking all the compounds. Do you have any idea what is causing this issue?
Thanks again.
Edit: I solve this issue by increasing the program_timeout time in all.ctrl file. Thanks
Hi Haroon,
great you solved the issue. I have program_timeout=9000. I guess it has to be higher than the total time of a job in vfvs-2 and slurm.
Best, Oliv