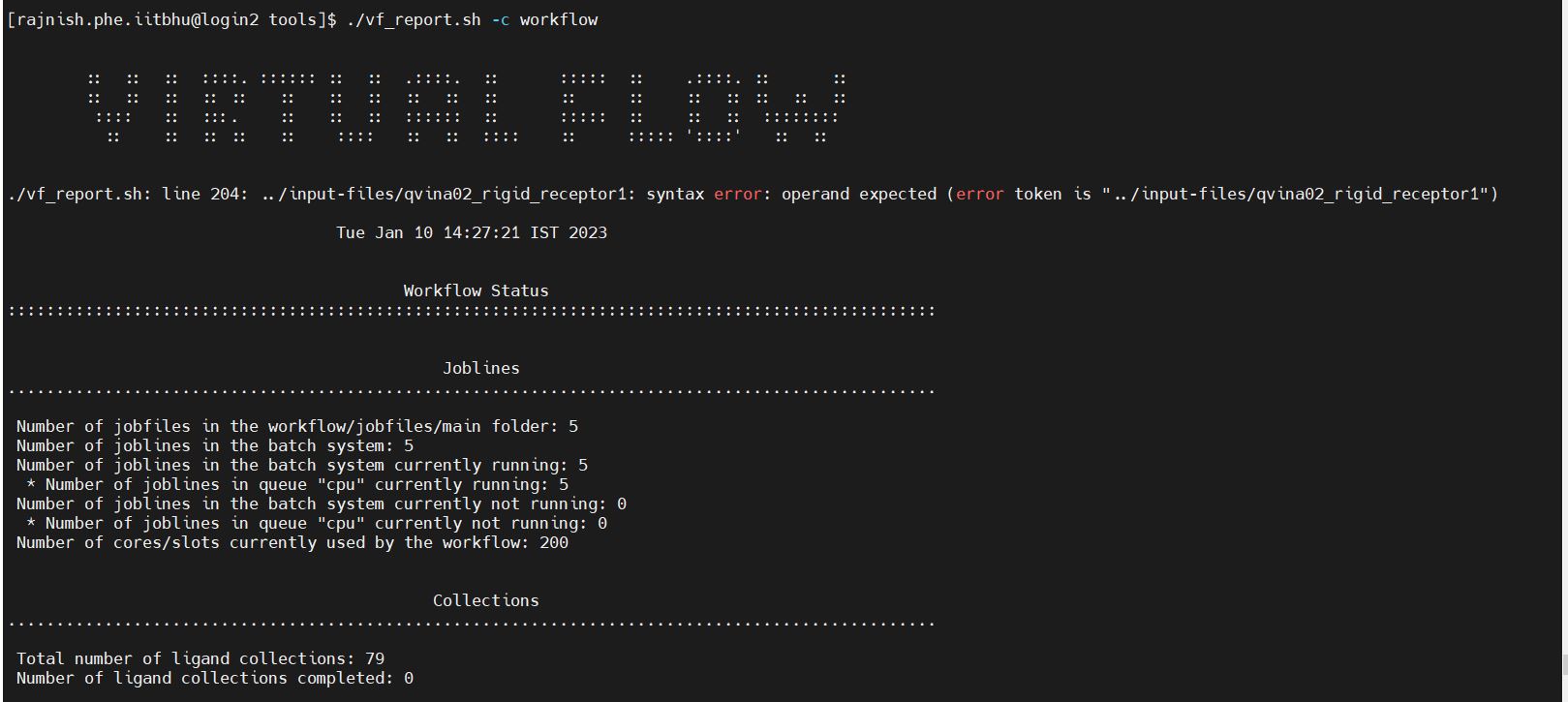
Getting the following error
./vf_report.sh: line 204: …/input-files/qvina02_rigid_receptor1: syntax error: operand expected (error token is “…/input-files/qvina02_rigid_receptor1”)
Please guide me how to solve the error and be able to successfully run the virtual screening
Hi @anurag ,
Welcome to the VF community!
Your error indicates it is related to line 204 in the file vf_report.sh, as the error message says.
If you look into the file vf_report.sh, you will find in line 204:
for value in ${docking_scenario_replicas_total[@]}; do
docking_runs_perligand=$((docking_runs_perligand+value))
done
This indicates that you might have some invalid settings in your all.ctrl file regarding the docking scenarios. Please check the values.
If you cannot find the problem, please paste the all.ctrl file here (the part with the docking scenarios). And in addition, you can change the code in the vf_report.sh file to:
for value in ${docking_scenario_replicas_total[@]}; do
echo $value
docking_runs_perligand=$((docking_runs_perligand+value))
done
that will print out the values and you will be able to see what is the invalid value.
Hope this helps,
Christoph
Thank you very much, I was able to solve the issue.
There is one more step that i need your help with.
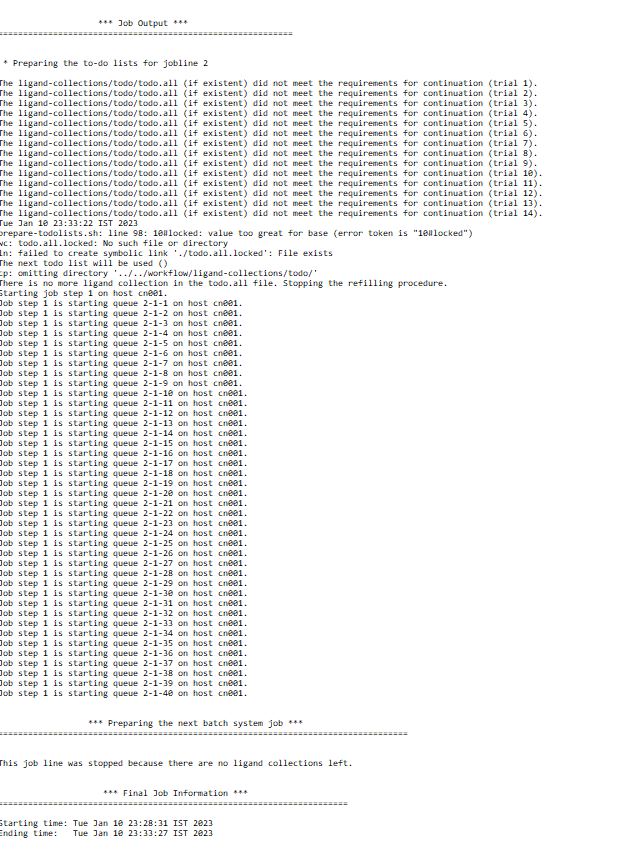
I am trying to run VFVS on a HPC system with parallelization for 50,000 molecules and tried to divide it into 5 parallel jobs having 1 node with 40 cores each the job initially submits and then one by one some of them just stops and finally at the end only one or two jobs are running and the job takes bit longer time to complete. So how do i get all the different submitted jobs to run in parallel without exiting and use the full potential and speed. Please guide me through the settings that I need to change in the scripts for the above.
Thank you for your time I really do appreciate it.
I’m glad you were able to solve the first problem.
Regarding your second problem, it sounds like there are some problems during the run. Did you try the troubleshooting methods pointed out in the documentation of VirtualFlow?
Best,
Christoph
Yes, I did try to understand the process better and now finally being able to successfully run.
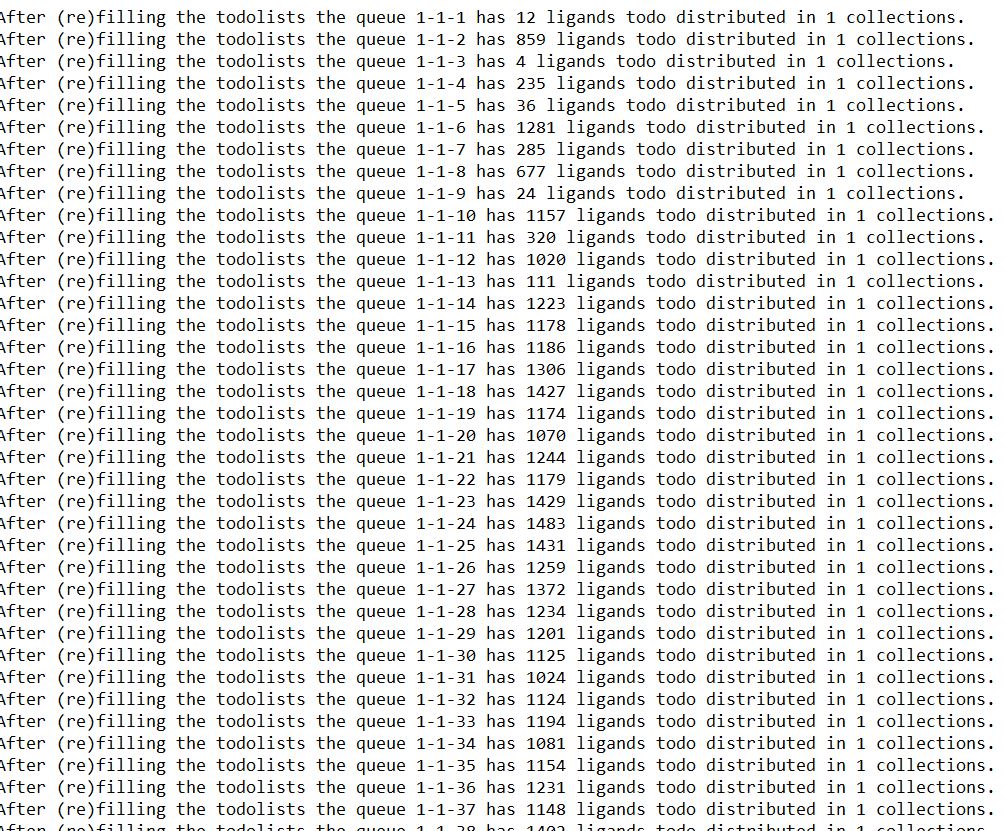
Can we make the ligand refilling distribution uniform in all the cores like all of them have same number of ligands to process?
I’m glad you were able to solve the problem ![]()
That is not possible with the bash version of VF, but will be in the future.