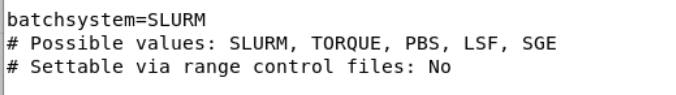
I have an additional question unrelated to the problem. In the provided example you used smina and qvina02, between the two files the only difference is the fact that for smina an additional scoring function is given (for both files the exhaustiveness is different). But what is the difference then durinig running? does one run use smina calculations and the other qvina02 to run the docking?
what would happen if i just use any protein with its own grid coordinates and gridsize (lets say prepared in vina and simply change the coordinates in the config file?
In other words, what if i prepare a protein in vina but tell virtual flow that it was prepared in qvina02 or smina? Since the config files are nearly identical
Syncing the jobfile of jobline 1 with the controlfile file …/…/workflow/control/all.ctrl.
Syncing the jobfile of jobline 2 with the controlfile file …/…/workflow/control/all.ctrl.
Syncing the jobfile of jobline 3 with the controlfile file …/…/workflow/control/all.ctrl.
Syncing the jobfile of jobline 4 with the controlfile file …/…/workflow/control/all.ctrl.
Syncing the jobfile of jobline 5 with the controlfile file …/…/workflow/control/all.ctrl.
submit.sh: line 68: sbatch: command not found
Error was trapped which is a nonstandard error.
Error in bash script submit.sh
Error on line 68
I’m also just trying to run tutorial 1 to test the program and become familiar. I have tried some of the suggestions posted here by Christoph without success. I have set the verbosity_logfiles=debug and store_queue_log_files=all_uncompressed
The workflow/outpu-files/jobs is also empty so I think i’m on the same boat as far as what Christoph mentioned:
" Since the workflow/output-files/queues folder is still empty as you said, the workflow seems to fail before starting the actual queues (one for each CPU) which process the ligands. Therefore also the workflow/ligand-collections/ligand-lists folder is still empty, since no ligands were yet processed."
So I might also not be reaching the queue.
I’m happy to provide any information that can help figure this mystery out.
I’m also relatively new with programming so I’m not so sure how to check how many nodes are available (that’s why I contacted the cluster admins for the settings info).
I have also tried the tutorial set up, without success:
i suggest switching queues per step and cpus per queue
but also check your squeue, maybe all nodes are used i noticed sometimes when it cannot get all at the same time it doesnt submit
I’ve been trying to change those but nothing seemed to changed. I saw in your threads that you were running an interactive session? I’m not so sure what this means but maybe I can look into it. Did that give you some information when you did it?
im not an exper in any ways so hence im just trying to figure out what is going wrong;
could you put # .bashrc under # source global definitions?
are you sure that there is atleast 1 node completely free with all of its cpu available? If for instance, 1 node with 32 cpu is available try steps_per_job=1 cpus_per_step=32 queues_per_step=32 cpus_per_queue=1
I understand, I really appreciate your suggestions and your help, and the fact the you are answering really fast. Unfortunately, I also did that last one change but nothing, I still got that error. I’ll keep looking into that and see if I can come up with something.
Ok, now it’s probably something I’m doing wrong because the error now it’s this one:
submit.sh: line 66: ./vf_start_jobline.sh: No such file or directory
Error was trapped which is a nonstandard error.
Error in bash script submit.sh
Error on line 66
This is how I entered the command but probably it’s not the right way.
Sorry for the very basic questions Do you have some pointers for this?
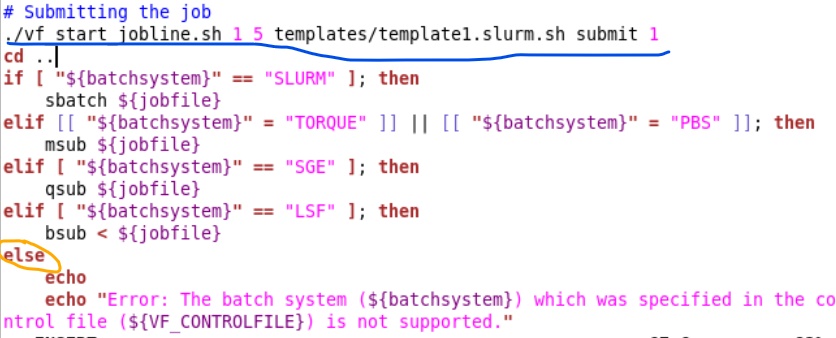
But I think that message you saw is just an “if” command (orange circle). Going back to your suggestion of adding the command myself. According to the errors, the system is trying to submit the job using the script “submit.sh” So I went to that file and I thought I could add the command in the section # Submitting the job. (underlined with the blue line in the picture) is that the correct way? and also, Is that the correct command?
Ah yes my previous statement was wrong, i thought that was a new error it gave you but it wase just the else part! Did you find the submit.sh file? I suggest trying to COPY it from /tools/helpers to /tools and then submit again and see what error you then get?
You could also try to make a copy and put it in tools/bin and then make sure you still have export PATH="/work/radiology/ID/VFVS-develop/tools/bin:$PATH"
if it doesnt work make sure you remove the copied files!
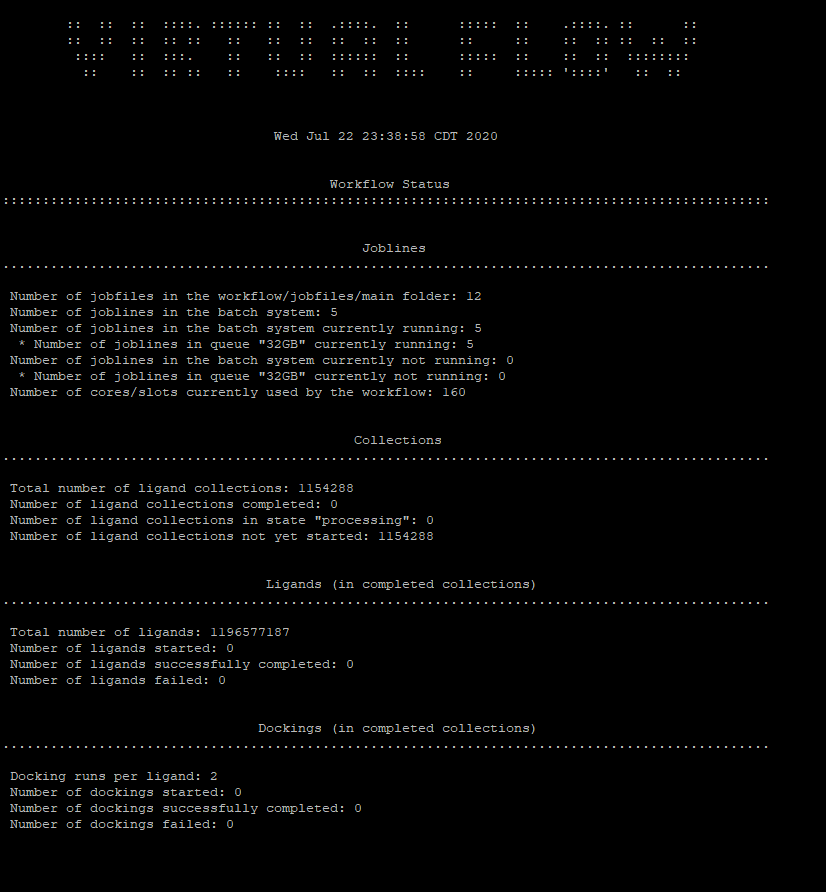
@kaneki good news! It seems to be running now. It turned out that for some reason my home domain in the cluster got erased but they fixed that. Now I’m able to submit jobs. This screenshot means it’s working correct?
I really appreciate all your help with this. Thank you!
This doesnt mean its working yet, it means it has been submitted. Once you see Number of dockings started and number of dockings completed you can consider it working
I see the options on this link but it would be helpful to see example commads on using them. It is not that clear to me as is. Right now, I am using “scancel -u username” but not sure if that cause problems for the consequtive runs after stopping.

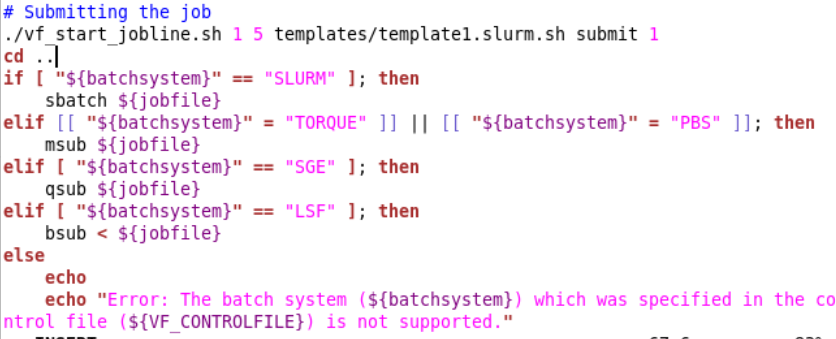
 Do you have some pointers for this?
Do you have some pointers for this?